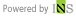KAIST, 인공지능으로 고성능 양자물성 계산시간 '획기적 단축'... "차세대 소재 설계의 새 시대를 열다"양자역학적 계산 속도, 인공지능으로 세계 최초 혁신 달성
연구팀이 개발한 새로운 방법론의 핵심은 3차원 화학 결합 정보를 예측하는 합성곱 인공신경망 딥러닝 모델인 '딥SCF(DeepSCF)'다. 이 모델은 슈퍼컴퓨터를 이용한 전통적인 양자역학적 계산에서 발생하는 복잡한 자기일관장(SCF) 과정을 인공지능을 활용해 대체한다. 기존 방식은 전자 밀도를 계산하고, 이를 기반으로 양자역학 방정식을 푸는 과정을 수십에서 수백 번 반복해야 했으나, 인공지능을 통해 이 과정을 단순화하고 시간 효율성을 크게 높였다.
밀도범함수론(DFT)은 양자역학을 통해 원자 단위에서 물질의 물성을 계산하는 대표적인 방법으로, 신소재 개발과 약물 설계 등 다양한 연구개발(R&D) 분야에서 표준적인 도구로 자리잡고 있다. 그러나 DFT 계산에서는 전자 밀도를 생성하고 이를 바탕으로 복잡한 양자역학 방정식을 수백 번 반복하는 SCF 과정을 거쳐야 한다. 이 때문에 수백~수천 개의 원자로 구성된 큰 시스템에는 계산 시간이 길어 적용에 어려움이 있었다.
김 교수 연구팀은 SCF 과정을 인공지능으로 대체할 수 있는지에 대해 연구하며, 전자밀도가 포함하는 양자역학적 정보를 인공지능이 학습할 수 있는 목표물로 설정했다. 전자밀도와 각 원자의 전자밀도 간의 차이를 나타내는 잔여 전자밀도(residual electron density)는 화학결합 정보를 포함하고 있어, 이를 바탕으로 딥러닝 모델에 학습을 시켰다. 연구팀은 다양한 화학 결합 특성을 지닌 유기 분자 데이터셋을 활용하여 모델이 다양한 분자 구조를 학습하도록 했다. 또한 회전 및 변형을 가해 일반화 성능을 높였다.
김용훈 교수는 “3차원 공간에 분포한 양자역학적 화학 결합 정보를 인공지능 신경망으로 예측하는 방법을 세계 최초로 구현했다”며, “이를 통해 전자구조 계산이 모든 스케일에서 물성 시뮬레이션의 기반으로 작용하게 될 것이며, AI를 통해 물질 계산을 획기적으로 가속화할 수 있다”고 말했다. 이번 연구는 AI와 물리학을 융합하여 고성능 전자구조 계산을 실현한 중요한 성과로 평가되며, 차세대 소재 및 소자 전산 설계에서 큰 역할을 할 것으로 기대된다.
이 연구는 KAIST 전기및전자공학부 김용훈 교수의 지도 하에 이룡규 박사과정 연구원이 제1저자로 참여한 결과로, 소재 계산 분야의 권위 있는 학술지 '네이쳐 파트너 저널 컴퓨테이셔널 머터리얼즈(Npj Computational Materials)'에 10월 24일자로 게재되었다. AI와 고성능 과학계산의 결합을 통한 혁신적인 성과로, 양자역학적 계산의 발전에 중요한 전환점을 제공하며 앞으로 다양한 R&D 분야에 걸쳐 응용될 가능성이 높다.
논문명은 Convolutional network learning of self-consistent electron density via grid-projected atomic fingerprints 이다.
이 기사 좋아요
<저작권자 ⓒ 특허뉴스 무단전재 및 재배포 금지>

댓글
KAIST, 인공지능, 양자역학 계산 가속화, 딥SCF 모델, 차세대 소재 설계 관련기사목록
|
많이 본 기사
사이언스 많이 본 기사
|























특허뉴스 소개 ㅣ 특허뉴스 조직도 ㅣ 개인정보취급방침 ㅣ 저작권보호 규약 ㅣ 이메일무단수집거부 ㅣ 광고안내 ㅣ 기사제보
한국특허신문사ㅣ제호: 특허뉴스 (月刊)ㅣ정기간행물 등록번호:서울강남 라01064ㅣ등록일: 2005년 4월6일
인터넷신문ㅣ제호: 특허TVㅣ등록번호:서울,아03336ㅣ등록·발행일: 2014년9월25일
발행소: 우(06132) 서울특별시 강남구 테헤란로 151(역삼동)ㅣ대표전화:02-2238-4345ㅣ팩스:02-2238-6769
발행/편집인: 이성용ㅣ청소년보호책임자: 이성용ㅣ뉴스제보: patentnews@naver.com
특허뉴스의 모든 컨텐츠(기사)는 저작권법의 보호를 받습니다. 무단 전재·복사·배포 등을 금지합니다.
Copyright ⓒ 2005 특허뉴스. All rights reserved.